TOP > ニュース > QBiCのこだわり > MetaBin:メタゲノム解析のための新しいツール

![]()
MetaBin: メタゲノム解析のための新しいツール
2012年6月26日
ゲノム科学と言えば、ヒトゲノム計画やヒトの身体を作る遺伝子についての研究を思い浮かべる人がほとんどだろう。これまでのゲノム研究の対象は遺伝子同士の関係や遺伝子とタンパク質との関係が主だった。しかし、今日、その対象は遺伝子と環境との関わりにまで広がっている。中でも環境中の微生物の遺伝子をコードするゲノムDNAを直接取り出して研究するメタゲノミクスと呼ばれる新しい分野が注目を集めている。従来、微生物を対象としたゲノム研究では、解析材料として十分な量のゲノムDNAを得るため実験室内で目的の細菌株を培養して増やし、そこからDNA を抽出した。しかし、大多数の細菌(約99%)は実験室内で培養すること自体が難しく、この従来型の方法で解析できる細菌の種類は限られている。一方、メタゲノミクス解析では培養を経ることなく、環境から採取した細菌の混合物をそのまま解析するので、すべての細菌についての情報を漏らさず得ることが出来る。環境中で微生物が形成するネットワークをそのまま理解することが出来るといえよう。一方、メタゲノミクスの試料に含まれる細菌種は非常に多く(数千種に及ぶこともある)、そこから、得られるDNA配列情報をもとに、どの細菌種が含まれているかを同定することは簡単な作業ではない。しかも、得られる配列データは断片的で、比較的少数しか含まれていない細菌種のDNA配列は正確に反映されないことも多い。皮肉なことに、DNA抽出やシーケンス技術が改良されることで取得可能なデータ量が増加し、メタゲノム解析により得られる多種由来の断片的な配列情報は膨大な量になっている(時として1012塩基対に上る)。
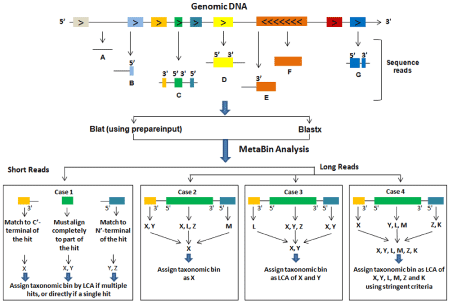
図1 オープンリーディングフレーム(ORF)ごとに高い相同性を有するゲノムを同定する。一つの配列データに複数のORF が含まれる場合、それらが同じゲノムにマッチした場合はその種を、複数のゲノムにマッチした場合はより上位の分類群(LCA)を付与する。
したがってメタゲノミクスの研究者が直面している課題の一つは、多種類のゲノム配列が入り混じったDNA断片群(いかに少量のデータであっても)がどういう細菌種(分類群:TAXON)に由来するかを高感度で検出するアルゴリズムを開発することだ。一方、アルゴリズムの感度を上げれば、通常解析に要する時間が長くなるが、それをどのように最小限に押さえるかが第二の課題だ。トッド・テイラーとメタシステム研究チームはMetaBin (Sharma et al. 2012)という新しい解析プログラムを開発した。これはこれまで、最も効率が良いとされていたMEGANという解析プログラムと同様にホモロジーサーチを使ったプログラムだが、MEGANと比べて同等か、あるいはより優れた性能を示した。特にメタゲノム解析では一般的な100ベース以下の短い配列情報の解析(図1)では、MEGAN の効率をはるかに凌駕した。
MetaBinの感度を最大化するための工夫の一つは配列データ中に見いだされるオープンリーディングフレーム(ORF)ごとにきめ細かく配列が由来する分類群を付与していくことと、同じDNA配列中に複数のORF が含まれる場合、すべてのORF がマッチする分類群(種あるいは属、科、門などのより上位の分類群)を付与することだ。これによってMEGAN よりも高い感度で分類群の同定が行える。新規ゲノムについても同様の性能を発揮する。また解析時間を短縮するために、MetaBin は BLAT(BlastXより高速)を使用する。これによって、時間効率を1000倍以上向上させることが出来た。
MetaBin の性能をMEGAN などの他の解析プログラムと比較するため、シミュレーションで混ぜ合わせた27種類の細菌のメタゲノムデータおよびヒトの腸内細菌叢から得られた既存のデータを用いて、配列群を属、科、門の3つの分類群に振り分けるテストを行ったところ、MetaBin は殆どの比較的長い配列群で、また全ての短い配列群で他の解析プログラムよりも精確に分類群の同定を行った。特に短い配列の解析における信頼性が高く、解析にかかる時間も大幅に短縮されていた。長い配列データの種レベルの分類群の同定においても MetaBin はMEGAN と比べて2倍の配列群に対して分類群を付与した。また、属、科、門のレベルでもMEGAN と同等の性能を示した。
テイラー博士は、MetaBin はメタゲノム解析における配列群が由来する細菌種・分類群同定に新たな方法を提供し、無数にある細菌の遺伝子やゲノムの構造及び機能の解明にまた一歩近づくことが出来たと考えている。「MetaBinの強みであるスピードと感度は誰しも求めるもので、今後、他のグループのメタゲノム解析パイプラインにも解析ツールの一つとして組み込まれて行くに違いない」と語る。
- Sharma VK, Kumar N, Prakash T, Taylor TD (2012) Fast and Accurate Taxonomic Assignments of Metagenomic Sequences Using MetaBin. PLoS ONE 7(4): e34030. doi:10.1371/journal.pone.0034030