TOP > News > Research Highlight > New Algorithms for Metagenomics

![]()
New Algorithms for Metagenomics
Most people when they hear the word “genomics” think of the human genome project and the mapping of every gene in the human body. Genomics, of course, has grown to be much more than that, as researchers are trying to understand the relationships a single gene has with not only other genes and proteins, but also the external environment. This is perhaps best reflected in metagenomics, an ambitious new field that studies bacterial genes and genomes taken directly from ecosystems. Normally in single bacterial genome projects, the species of interest are grown in a laboratory to obtain enough DNA for sequencing. However, this limits the number of bacteria one can study dramatically, as the vast majority cannot be cultured in the lab. Metagenomics, by completely avoiding the culturing step, has the potential to access 100% of the information in any given environment. By studying a more comprehensive set of bacteria, which can range from just a few different species to several thousands, scientists can make better conclusions about the networks in which the bacteria function and thrive. However, because the typical metagenomic sample consists of a mixture of many different bacterial species, it is difficult to reassemble the individual bacterial genomes and to assign the sequence reads and their predicted genes back to the species of origin. Additionally, the sequences are fragmented and may not completely represent the whole environment, especially in the case of minor species within the environment. Ironically, the heterogeneous and fragmented genetic information resulting from metagenomic sequencing has resulted in more data, sometimes reaching Terabases (1012 base pairs), as better extraction and sequencing methods mean more genetic information can be acquired, even if in many cases the genetic data is only piecemeal.
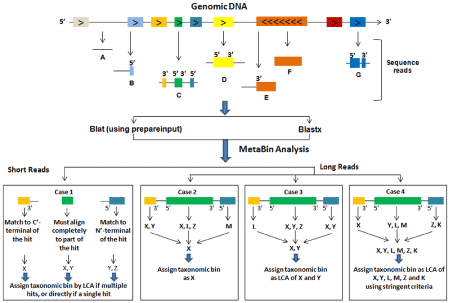
Figure 1. ORF-based approach for the taxonomic assignment of reads of different lengths derived from different regions of the genomic DNA.
This creates two paradoxal challenges for informaticians studying metagenomic data. One is to increase the sensitivity of their algorithms so that they can accurately assign genes to their appropriate taxon even when the data itself is marginal. The second, however, is to minimize the time required for this analysis, which often comes at the expense of sensitivity. Todd Taylor and his team at the Laboratory for MetaSystems Research feel they have made a significant gain in dealing with these juxtaposed problems with a new homology-based program, MetaBin (Sharma et al. 2012), that matches or surpasses the performance of its closest rival, MEGAN, when examining long reads, but outperforms them with regards to shorter ones, especially those less than 100 bp, a common circumstance in metagenomics (Figure 1).
To optimize MetaBin’s sensitivity, the group considered the possible outcomes of taxonomy assignment for a given read from the taxonomic information of all ORFs in the sequence to assign the sequence to its most appropriate taxonomic bin. The sequence is first aligned against a reference database and then its taxon is assigned based on the most significant matches (protein hits). MetaBin assumes that the correct genome hit should match all the ORFs, which results in a higher sensitivity than that offered in MEGAN. This is even true for novel genomes. To decrease time requirements, MetaBin was made compatible with BLAT, thus improving the time efficiency by three orders of magnitude.
The group tested their program using simulated read datasets from 27 different microbial genomes and published human gut metagenomic data and compared its performance to other programs, including MEGAN, when assigning sequences to three hierarchical levels – genus, family, and phylum. For most long read comparisons and all short read comparisons, MetaBin outperformed all of the other tested programs. It especially improved sensitivity for short reads and dramatically increased the speed that punctuates MetaBin’s usefulness.
Depending on the metagenomic environment, Metabin failed to match a large number of reads in the NR database. However, this makes it no more limited than current models and when considering its effectiveness for the analysis of short reads and computational time savings makes MetaBin the option of choice. Furthermore, when it came to the analysis of long reads, MetaBin was able to assign twice the number of reads at the species level than MEGAN while performing equally at the phylum, family, and genus levels.
Dr. Taylor believes that the advances in MetaBin offer an alternative approach to metagenomic taxonomic analysis that will bring us closer to understanding the composition and function of innumerable bacterial genes and genomes. “Given MetaBin’s speed and sensitivity there is really no reason why any group would not want to use it or include it in their own metagenomic analysis pipelines,” quotes Todd.
- Sharma VK, Kumar N, Prakash T, Taylor TD (2012) Fast and Accurate Taxonomic Assignments of Metagenomic Sequences Using MetaBin. PLoS ONE 7(4): e34030. doi:10.1371/journal.pone.0034030